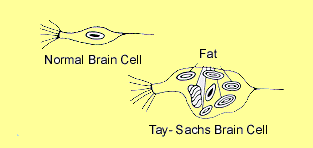
TAY-SACHS DISEASE
![]()
|
|
TAY-SACHS DISEASE
![]()
|
|
![]() FOR FURTHER INFORMATION
FOR FURTHER INFORMATION
Desnick, Robert J. and Michael M. Kaback (eds.). Tay Sachs Disease. Academic Press, 2001.
Scriver, Charles et al. Metabolic and Molecular Bases of Inherited Disease, 7th ed., McGraw Hill Inc.,1995.
![]() STUDIES ON CARRIER POPULATIONS
STUDIES ON CARRIER POPULATIONS
1998. "Carrier Screening for Cystic Fibrosis, Gaucher Disease, and Tay-Sachs Disease in the Ashkenazi Jewish Population: The first 1000 cases at New York University Medical Center, New York, NY." David Kronn et al., Arch Intern Med, vol. 158: 777-781.
1996. "Twenty-Year Outcome Analysis of Genetic Screening Programs for Tay-Sachs and ß-Thalassemia Disease Carriers in High Schools." John Mitchell et al., American Journal of Human Genetics, vol. 59: 793-798.
1996. "Heterozygosity for Tay-Sachs Disease in non-Jewish Americans with ancestry from Ireland or Great Britain." Margaret van Bael et al., J Med Genet, vol. 33: 829-832. *This is the study that finds TSD rates for Irish Americans fall between 1 in 192 to 1 in 52.
1993. "Tay Sachs Disease - Carrier Screening, Prenatal Diagnosis, and the Molecular Era: An International Perspective, 1970 to 1993." Michael Kaback et al., JAMA Vol. 270, no. 19: 2307-2315. *The most comprehensive study that I have come across to date. Data collected through University of California, San Diego, from nearly 1,000,000 young adults of both Jewish and non-Jewish ancestry.
![]() OTHER FORMS OF TAY-SACHS DISEASE
OTHER FORMS OF TAY-SACHS DISEASE
LOTS - Late Onset Tay Sachs Disease: tel. 1-800-672-2022
Tay-Sachs has three clinical forms: infantile, juvenile, and Late-Onset Tay-Sachs (LOTS). The juvenile and adult Late-Onset forms are much less common than the infantile form. Unlike the infantile form where there is no enzyme, some Hex-A enzyme activity is present in both juvenile and late-onset, but the levels are very low. Unfortunately, there is the same progressive deterioration of central nervous system, just at different ages.
![]()